
关注泰科科技 做模拟不迷路

摘要
—www.tech-box.com.cn—
作者用耦合团簇量子化学方法计算了从氢分子结构中切割出的分子团簇的能量学,该结构模拟了观察到的固体氢相。通过Kohn-Sham密度泛函理论(DFT)计算得到了在150、250和350 GPa压力下的氢结构,在该压力范围内II、III和IV相是稳定的。报告了不同泛函的耦合聚类数据的DFT能量的计算偏差,并生成了优化的泛函,减小了误差。作者给出了半局域和杂化密度泛函的建议,期望它们能准确描述高压下的氢。
引言
—www.tech-box.com.cn—
固体氢的相图具有重要的理论和实验意义。例如,迪亚斯和西尔维拉最近的实验声称强压缩的氢会金属化,这引起了全世界的关注和争议。目前已经采用了密度泛函理论(DFT)、扩散量子蒙特卡罗(DMC)和路径积分分子动力学(PIMD)等方法来研究氢的结构和能量。在这里,作者构建了交换相关泛函,有望在DFT中提供上述结构的能量学的准确描述。提供了两个精确的混合泛函,分别表示为O1和O1- d3,用于没有和有色散修正。作者还提供了精确的广义梯度逼近(GGA)泛函(尽管不如它们的混合对应函数精确),分别表示为O3和O3- d3,用于不进行色散修正和有色散修正。利用其中一个泛函生成的轨道计算DMC能量,并与耦合单簇、双簇和扰动三簇[CCSD(T)]的结果进行比较,以便进一步评估氢泛函的准确性。
计算方法
—www.tech-box.com.cn—
作者使用小分子氢团簇作为参考系统来构建作者的DFT泛函。作者从稳定的块状DFT结构中提取团簇几何形状,该结构是由PBE半局域密度泛函得到的,通过对每个包含24个氢原子的系统进行球形切割得到的。在CCSD(T)和DFT水平上对各种氢分子簇的总能量进行了计算。耦合集群和DFT计算是使用MOLPRO代码执行的。
结果与讨论
—www.tech-box.com.cn—
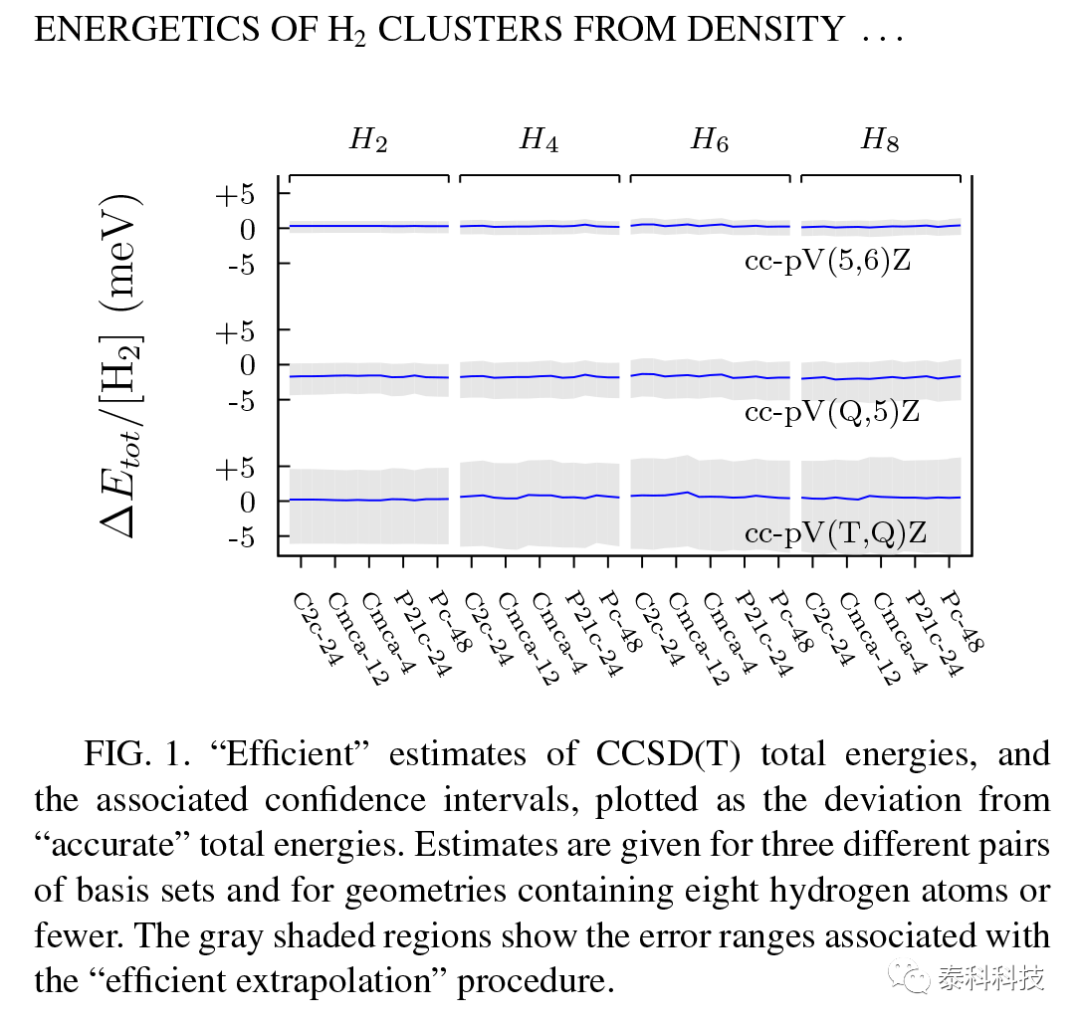
对于N=2-8的小簇子集,该高效程序提供的总能量与精确值一致,在估计误差范围内。高效(TQ)、(Q5)和(56)外推的平均绝对误差分别为6.1、2.6和1.0meV/[H2],其中作者使用符号(n1n2)表示cc-pVn1Z和cc-pVn2Z基组结果的有效外推。有效估计的能量和每个小簇几何的置信区间显示在图1中,作为精确估计的偏差。
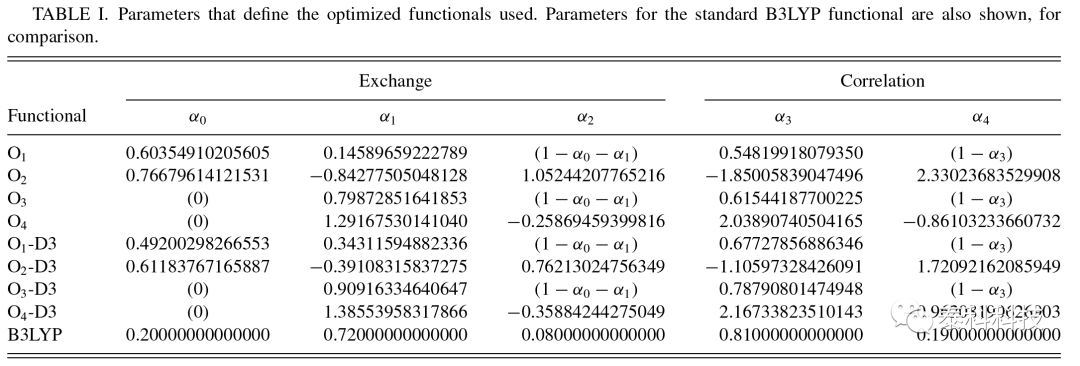
8种泛函都具有较快的收敛速度,4次迭代后总能量的变化均小于10-3meV/[H2]。表I列出了更优参数和约束条件。
作者测试了有和没有色散校正的泛函,提供了总共30个不同的DFT哈密顿量(见表II)。
每个泛函的误差汇总为图2中具有和不具有色散修正的泛函的所有聚类的平均值和更大绝对差。
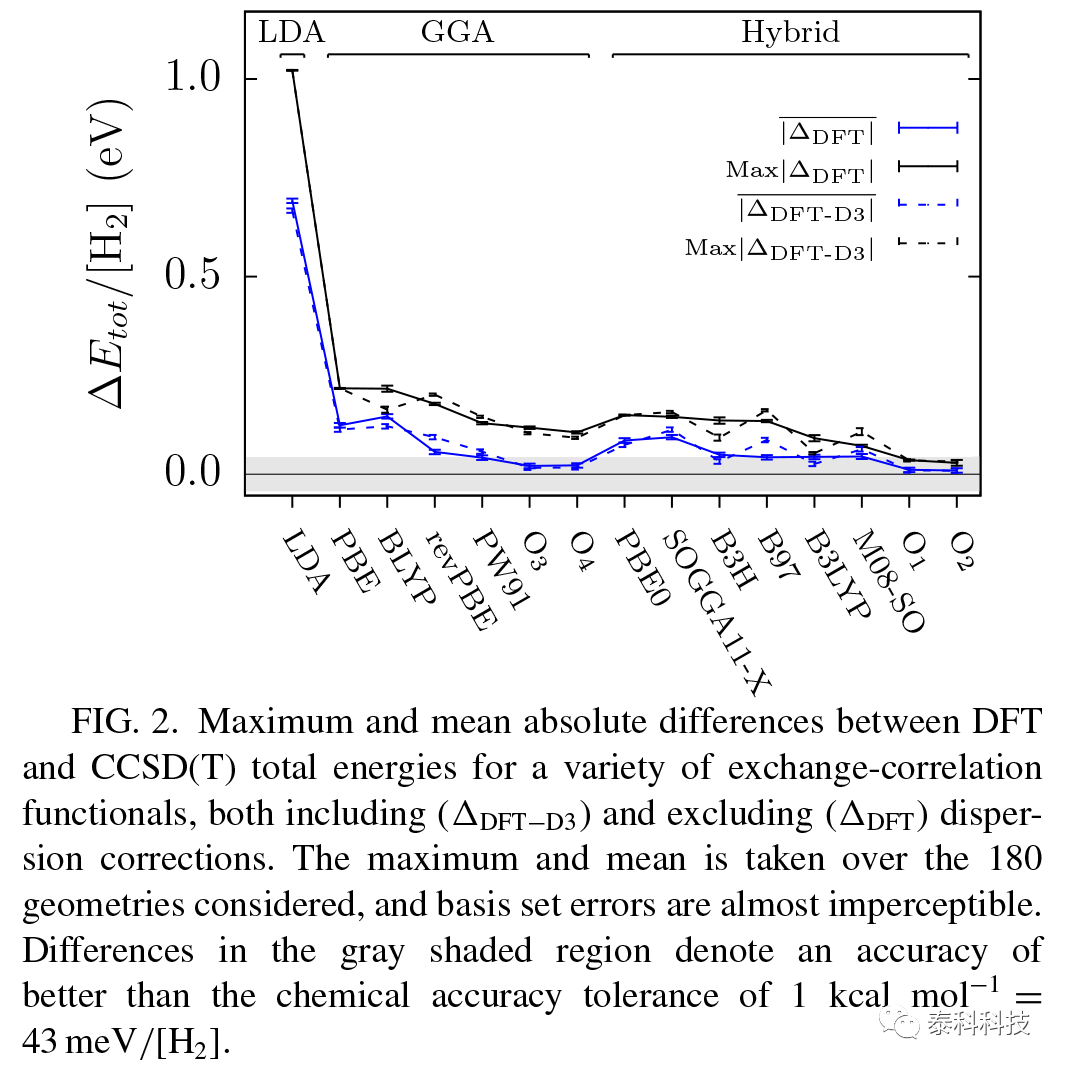
图3显示了陡峭的分段(近似)线性变化与下面的大块结构的压力,其中每个分段与不同的对称性和簇大小相关。对于杂化泛函,这种依赖并不明显。
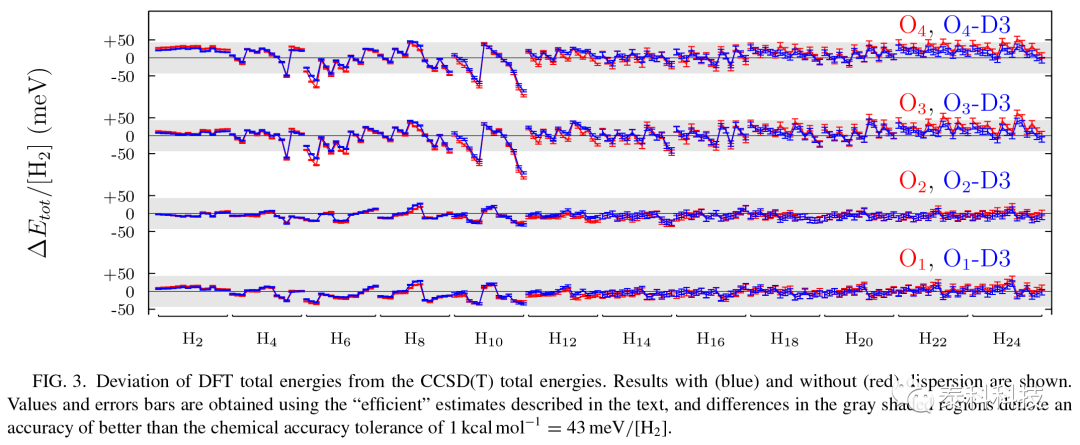
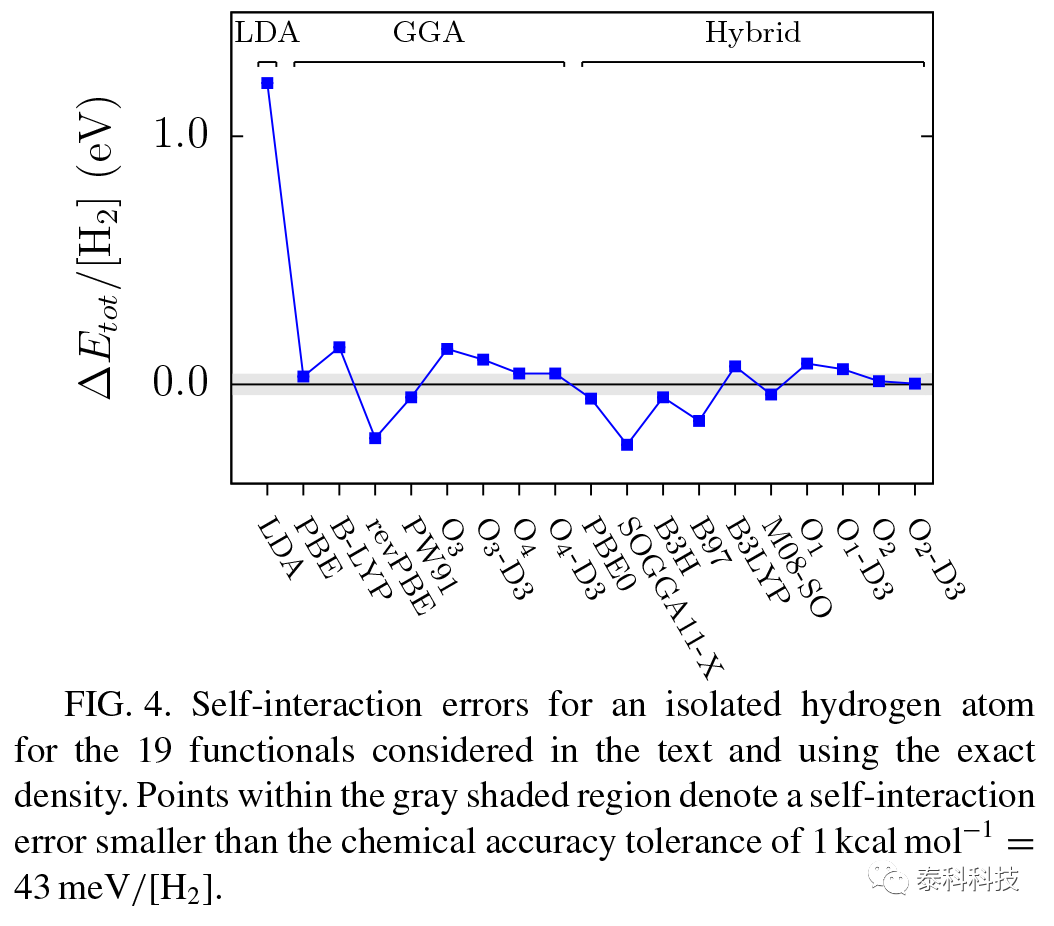
图4显示了每个泛函的SI误差(对于一个孤立的原子弥散为零,因此只有19个不同的总能量需要考虑)。更大的SI误差出现在LDA函数。对于更佳的官能团,通过加入HF交换和不强制HEG限制,SI误差持续降低。在所有考虑的泛函中,o2和do2 – d3杂化泛函的SI误差最小,分别为13和3meV/[H2]。
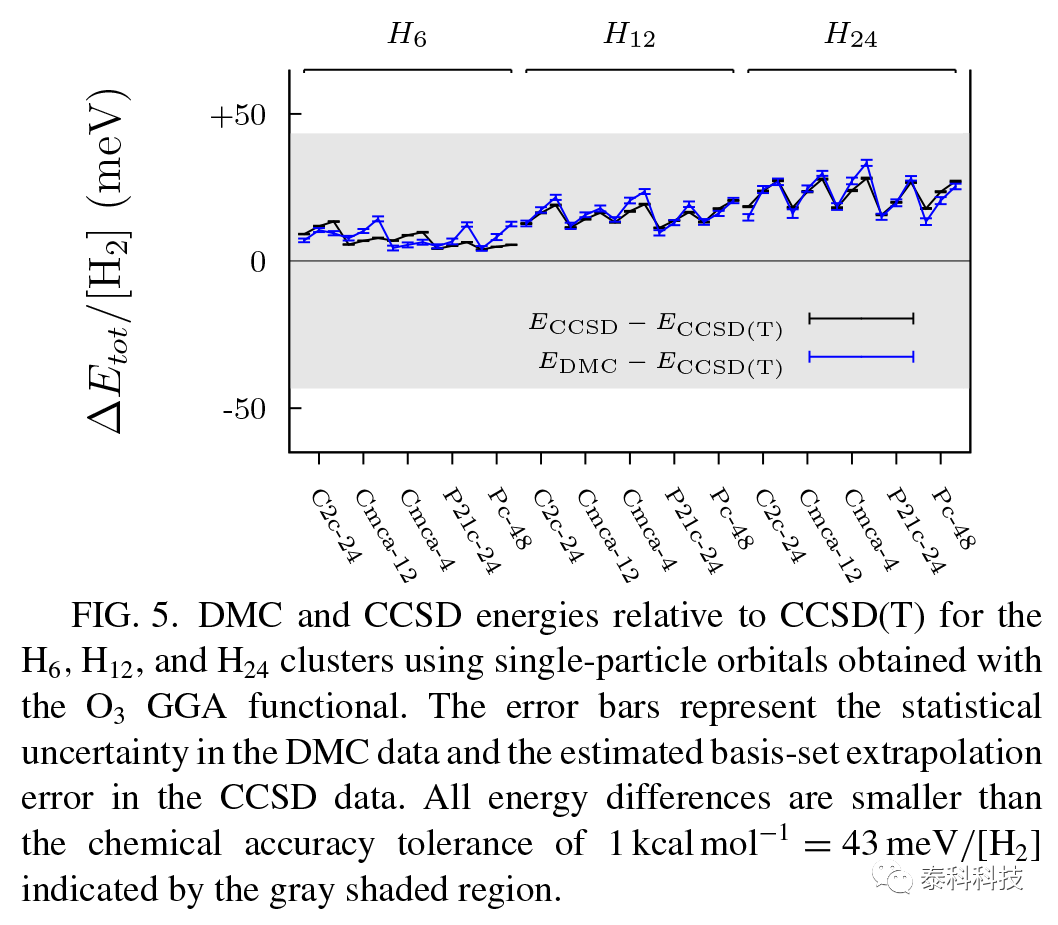
在图5中,作者绘制了对应于O3泛函的DMC能量, 图5还显示了DMC和CCSD能量之间的密切一致,强烈表明作者的DMC结果中缺失的小的相关能可以被CCSD(T)中包含的三个激励所识别。
总结
—www.tech-box.com.cn—
也许改进泛函的最有希望的选择是为优化的泛函添加更多的变分自由度。最简单的方法是包含半局部泛函的其他参数化,包括元项,因为这将保持作者在迭代优化过程中依赖的优化的线性。更通用的方法是直接优化函数形式中的增强因子,在每个优化迭代中引入非线性。类似地,在色散校正中出现的筛选参数也可以得到优化,但这似乎只能对能量的精度提供边际改进。

公司简介
—www.tech-box.com.cn—
北京泰科博思科技有限公司(Beijing Tech-Box S&T Co. Ltd.)成立于2007年,是国内领先的分子模拟及虚拟仿真综合解决方案提供商。
北京泰科博思科技有限公司与国际领先的模拟软件厂商、开发团队深入合作,为高校、科研院所和企业在材料、化工、药物、生命科学、环境、人工智能及数据挖掘、虚拟仿真教学等领域提供专业的整体解决方案。用户根据需要在我们的平台上高效的进行各种模拟实验,指导实际的生产设计。
北京泰科博思科技有限公司拥有一支一流的技术服务团队和资深的专家咨询团队,以客户真正需求出发,服务客户,为客户创造价值。我们秉承“职业、敬业、担当、拼搏、合作”的企业精神,致力于用国际领先的软件产品和专业全面的技术支持服务,成为客户可信赖的合作伙伴。